Package Overview
This package aims to extract meaningful features from a protein's 3D structure.
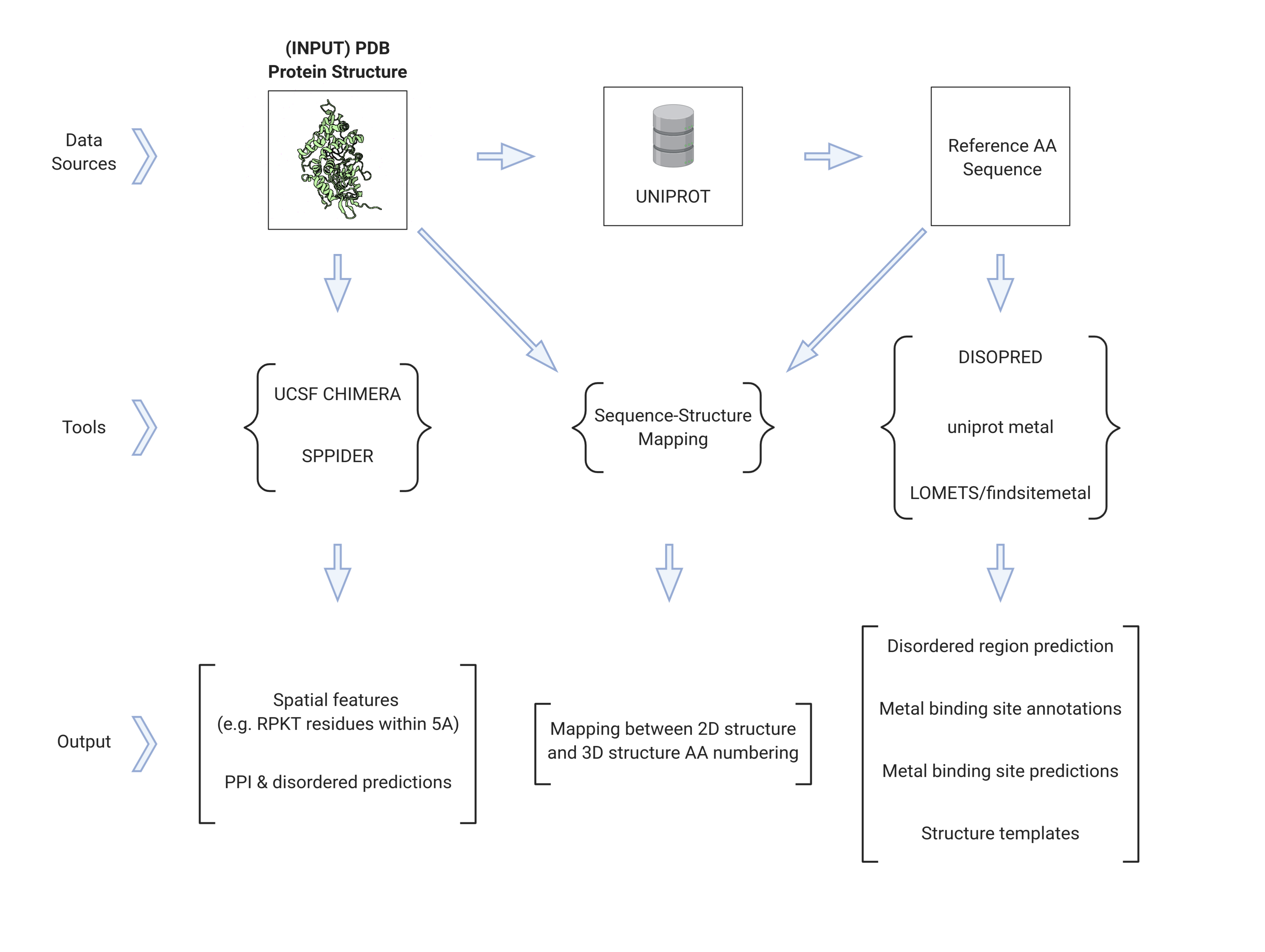
When you have a 3D structure, there are various different tools that you can use to extract different features. This package packages together those tools, along with some additional helper scripts.
Below, I'll upload a graphical representation of how all of the tools in the ProteinFeatures repository connect. Then, I'll give a quick blurb about each.
Pre-requisites: generating a 3D protein structure
Before extracting any features, you will need a 3D structure. Or, if you only have a 3D structure, you will need the 'canonical' reference sequence for the protein represented in your 3D structure.
To get a 3D structure, you can use a modelling software (we use I-TASSER and/or QUARK or MODELLER) or, if available, you can use an available structure from the PDB.
For info on that, see the Prerequisities section.
Chimera
UCSF Chimera is a GUI-based software, with a built-in Python 2.7 interpreter, that can load and manipulate PDB structures.
The bulk of the work done in this repository has been building a pipeline to extract features from RPKT residues using Chimera. This is a bit tricky, since some of the features we need seem to only be available via the GUI and via Chimera's (somewhat outdated) interpreter.
For more info on using the pipeline, see the Chimera section.
LOMETS
LOMETS is a tool in the I-TASSER suite that, given an amino acid sequence, will search different protein structure databases to try to find homologous templates. In the I-TASSER suite, these templates are used for threading. In this feature extraction package, they are used as input to findsitemetal.
For more info, see the LOMETS section.
Finding metal binding sites
Metal binding sites can be predicted, via findsitemetal. Some structures also have experimentally-determined metal binding sites, which you can querey from uniprot.
For more info, see the Metal Binding section.
SPPIDER
SPPIDER is an online tool that, given a 3D structure (in PDB format) predicts metal binding sites and disordered regions.
For more info, see the SPPIDER section.
DISOPRED
DISOPRED is a command line tool that, given a 2D amino acid sequence (in FASTA format), predicts disordered regions.
For more info, see the DISOPRED section.
Sequence-Structure mapping
Oftentimes, the residue numbering in a PDB file is not the same as the residue numbering in FASTA sequence of the associated canonical sequence. So, I wrote a short pipeline to map between a 3D and 2D structure.
For more info, see the Sequence-Structure Mapping section.